СТАЙЛАБ предлагает тест-системы для определения левомицетина (хлорамфеникола) в молоке в соответствии с ГОСТ Р 52842-2007 (ИСО 18330:2003), сухом молоке, масле, сыре, твороге, молочных продуках (кефире, сметане, йогурте, йогурте с фруктами), меде, пчелином маточном молочке, креветках, рыбной муке, мясе, яйцах по МУК 4.1.3535-18, сыворотке крови/плазме, моче, комбикормах, ферментах, вине и виноградном соке.
Иммунохроматографический метод анализа, тест-полоски
9268 Сухое молоко с содержанием хлорамфеникола 0,13 мкг/кг, 17 мл
9267 Сухое молоко с содержанием хлорамфеникола 0,34 мкг/кг, 17 мл
9269 Сухое молоко с содержанием хлорамфеникола менее 0,015 мкг/кг, 17 мл, для отрицательного контроля
S-4032 cтандарт хлорамфеникола SPEX
Чистые вещества и стандарты сульфаниламидов, нитроимидазолов, пенициллинов, амфениколов для анализа в соответствии с ГОСТ 54904-2012
Левомицетин (хлорамфеникол, хлоромицетин) – это антибиотик широкого спектра действия. Он обладает бактериостатическим действием и эффективен против возбудителей таких заболеваний, как дизентерия, брюшной тиф, пневмония, газовая гангрена, сибирская язва бактерий рода Yersinia (возбудители иерсиниоза, псевдотуберкулеза и чумы) и многих других опасных микроорганизмов. Хлорамфеникол очень хорошо растворяется в жирах и в значительных количествах выделяется с молоком. Его молекула имеет небольшие размеры, и это, вместе с жирорастворимостью, позволяет ему легко проникать во все ткани тела, в том числе, в мозг.
ВОЗ рекомендует использовать масляную суспензию хлорамфеникола в качестве первой линии терапии менингита в странах с низким уровнем дохода. В форме мазей для наружного использования он применяется при фурункулезе и других кожных заболеваниях, а также при ранениях. Многие лечат с помощью левомицетина пищевые токсикоинфекции («пищевые отравления»). Согласно источникам, хлорамфеникол проявляет активность в отношении некоторых крупных вирусов. При этом микроорганизмы очень медленно вырабатывают резистентность к нему, потому левомицетин остается очень эффективным антибиотиком.
В сельском хозяйстве и ветеринарии левомицетин применяется для лечения животных и птицы, больных желудочно-кишечными заболеваниями, в том числе, кокцидиозом, который вызывают паразиты. Также его используют для лечения заболеваний дыхательных путей.
Впервые хлорамфеникол выделили в 1947 году из культуры почвенного микроорганизма Streptomyces venezuelae. В клинической практике он используется с 1949 года. Позднее левомицетин научились синтезировать искусственным путем, и в настоящее время его получают именно таким способом.
В России хлорамфеникол входит в список жизненно необходимых и важнейших лекарственных препаратов и применяется в животноводстве и ветеринарии. В США левомицетин не выпускается для перорального употребления с 1991 года. В странах Евросоюза использование этого средства в животноводстве запрещено. Рекомендации применять хлорамфеникол только для лечения тяжелых заболеваний, запреты на его использование в животноводстве или необходимость контролировать содержание левомицетина в пищевых продуктах связаны с побочными эффектами этого средства, возникающими при хроническом воздействии на человека. Одним из таких эффектов являются реакции гиперчувствительности вплоть до анафилактического шока. Но чаще они выражаются в крапивнице и других кожных проявлениях, особенно при использовании мазей с левомицетином.
Левомицетин метаболизируется в печени с образованием глюкоронида хлорамфеникола – неактивного соединения. В этом виде он выводится почками. Хлорамфеникол ингибирует активность одного из ферментов печени, участвующего в обмене стероидов и некоторых других соединений, а также в нейтрализации токсичных веществ, что нарушает работу печени. В некоторых случаях левомицетин вызывать почечные кровотечения. Хлорамфеникол долгое время остается в организме животных и в пищевых продуктах, в том числе, в мясе и молоке.
Длительное употребление левомицетина в высоких дозировках может вызывать желудочно-кишечные расстройства, раздражение слизистых оболочек рта и желудочно-кишечного тракта, а также возникновение язв на них. Кроме того, он подавляет микрофлору кишечника, что часто приводит к расстройствам пищеварения или вторичной грибковой инфекции.
Самые опасные эффекты хронического употребления левомицетина связаны с его воздействием на кроветворную систему. Он вызывает апластическую анемию (атрофию кроветворения): неизлечимое заболевание, вызванное повреждением клеток костного мозга. При ней снижается количество всех трех типов клеток крови: лейкоцитов, эритроцитов и тромбоцитов. Это редкое осложнение, и при употреблении левомицетина оно чаще всего возникает, если это средство применяли перорально. К другим осложнениям со стороны кроветворной системы относится подавление работы костного мозга, а также лейкемия («рак крови»). Чаще всего такая реакция возникает у детей, причем риск ее возникновения увеличивается с увеличением длительности употребления левомицетина.
Дозы левомицетина, способные вызывать побочные эффекты, в том числе, апластическую анемию, до сих пор не определены. Поэтому в странах Евросоюза использование левомицетина при производстве пищевых продуктов животного происхождения запрещено. Минимальный предел чувствительности методов анализа, с помощью которых в Евросоюзе допускается проводить определение левомицетина (хлорамфеникола) в пище составляет 0,3 мкг/кг.
Согласно гигиеническим требованиям к качеству и безопасности продовольственного сырья и пищевых продуктов, принятым в Российской Федерации и странах Таможенного Союза, в соответствии с Техническим Регламентом Таможенного Союза ТР ТС 021/2011 («О безопасности пищевой продукции») содержание левомицетина (хлорамфеникола) в яйцах, мясе и других продуктов не допускается (должно быть менее 0,01 мг/кг, или 10 мкг/кг). Такие же нормы установлены для молочных продуктов Техническим Регламентом Таможенного Союза ТР ТС 033/2013 «О безопасности молока и молочной продукции». С 1 июля 2015 года, согласно ТР ТС 033/2013, максимально допустимое содержание левомицетина в молоке не должно превышать 0,0003 мг/кг (0,3 мкг/кг), что соответствует также нормам Евросоюза. С актуальными законодательными нормативами можно ознакомиться на сайте compact24.com.
Остаточные количества левомицетина (хлорамфеникола) в пищевых продуктах можно определять методами радиоиммуного анализа и жидкостной хроматографии высокого давления. Однако данные методы реализуются на дорогостоящем оборудовании и занимают длительное время. Кроме того, радиоиммунный метод анализа предполагает использование радиоактивных материалов, а хроматографические методы требуют квалифицированного обслуживания и трудоемки. Микробиологические методы определения антибиотиков, в том числе, левомицетина, просты и дешевы, но недостаточно специфичны, поскольку многие микроорганизмы чувствительны к различным антибиотикам.
Для скрининга и в рутинной лабораторной практике широко применяют иммуноферментный метод анализа. Например, с сентября 1998 г. в Германии ИФА является официальным методом скрининга остатков левомицетина в молоке; см.§ 35 LMBG, 01.00 68. В Белоруссии утверждены МУК 10-1-5/118/В от 18.08.2003 по контролю левомицетина в молоке, сухом молоке, яйцах и мясе с помощью тест-системы RIDASCREEN® Chloramphenicol. В России метод иммуноферментного анализа для определения левомицетина в пищевых продуктах утвержден МУК 4.1.1912-04.
источник
Изобретение относится к ветеринарной токсикологии и может быть использовано при определении содержания левомицетина в кормах животного происхождения.
Известен способ количественного определения левомицетина в пищевых продуктах и фармпрепаратах, заключающийся в том, что левомицетин переводят из пробы в раствор, проводят кислотный гидролиз и осаждают белок из гидролизата с последующим вольтамперометрическим определением, вольтамперометрическое определение левомицетина осуществляют путем регистрации катодных пиков антибиотика на индикаторном ртутно-пленочном или стеклоуглеродном электроде в дифференциальном режиме съемки вольтамперограмм при соответствующих потенциалах -(0,67±0,05) В и -(0,60±0,03) В относительно насыщенного хлорид-серебряного электрода на фонах 0,1 моль/дм 3 аммония лимоннокислого двузамещенного (рН 4,7-5,1) или 0,1 моль/дм 3 (NH4)2SO4 с добавлением НСl до рН 5,1 при скорости развертки потенциала 10-25 мВ/с и концентрацию левомицетина определяют по высоте пика методом добавок аттестованных смесей [Патент RU №2180748, МПК G01N 27/48, 20.03.2002].
Недостатками известного способа являются низкая чувствительность и длительность проведения анализа.
Наиболее близким по технической сущности к заявляемому способу является определение остаточных количеств левомицетина (Хлорамфеникола, Хлормицетина) в продуктах животного происхождения методом высокоэффективной жидкостной хроматографии, который заключается в отборе проб — 10 г мяса, гомогенизации с 15 см 3 фосфатного буфера (рН=6,88), последующей экстракции 3 раза по 30 см 3 этилацетата. Полученную взвесь центрифугируют 15 мин при 4000 об/мин и декантируют. Этилацетатный слой промывают последовательно 5 см 3 насыщенного раствора NaCl с добавлением 0,2 мл 10% NaOH; 5 см 3 насыщенного раствора NaCl с добавлением 0,2 см 3 10%-ного СН3СООН; 5 см 3 насыщенного раствора NaCl. Органический слой отбирают и упаривают на ротационном испарителе (при 50°С) до возможно минимального объема, отдувают азотом до исчезновения запаха органических растворителей, добавляют 3 см 3 смеси ацетонитрил-вода (1:4) и экстрагируют 3 раза по 5 см 3 петролейным эфиром. Петролейный эфир отбрасывают и извлекают левомицетин экстракцией этилацетатом (3 раза по 5 см 3 ). Этилацетатный слой упаривают досуха, растворяют в 0,1 см 3 метанола. В жидкостной хроматограф с колонкой (Ультрасфер ODS, C18) 5 мкм (250×4,6 мм), УФ-детектором при λ=278 нм, используя элюент ацетонитрил-вода-дециламин (40:60:0,1), вводят 5 мкл стандартного раствора (10 нг/мкл) левомицетина. В тех же условиях вводят метанольный экстракт образца мяса. Проводят обработку результатов. За окончательный результат измерения принимают среднее арифметическое результатов двух параллельных определений. [Определение остаточных количеств левомицетина (Хлорамфеникола, Хлормицетина) в продуктах животного происхождения методом высокоэффективной жидкостной хроматографии и иммуноферментного анализа. Методические указания. МУК 4.1.1912 — 04 // Сборник методических документов, необходимых для обеспечения применения Федерального закона от 12 июня 2008 г. — №88 ФЗ «Технический регламент на молоко и молочную продукцию». — М.: Федеральный центр гигиены и эпидемиологии Роспотребнадзора, 2009. — С. 41-47
Недостатками данного метода определения левомицетина является его низкая чувствительность и избирательность, длительность проведения анализа.
Техническим результатом предлагаемого изобретения является повышение чувствительности и избирательности способа, сокращение длительности проведения анализа.
Технический результат достигается тем, что в способе определения левомицетина в кормах животного происхождения с использованием высокоэффективной жидкостной хроматографии, включающем отбор пробы, гомогенизацию, экстракцию, упаривание, растворение сухого остатка, введение растворенного сухого остатка в хроматограф с использованием элюента, обработку результатов анализа, в качестве пробы берут навеску кормов животного происхождения массой от 200 до 500 мг, экстракцию проводят 95% этанолом, перед упариванием экстракт фильтруют, обезвоживают сернокислым натрием, растворение сухого остатка проводят в этилацетате, в качестве хроматографа используют жидкостной хроматограф с детектором спектрофотометрическим UVV 104М и колонкой Диасфер-110С-16 (150×4) мм, с размером частиц сорбента 5 мкм, в качестве элюента используют смесь ацетонитрил-вода-диэтиламин в соотношении 30:70:0,1.
Уменьшение массы пробы корма животного происхождения позволяет уменьшить объем экстрагента и количество извлекаемых при экстракции коэкстрактивных веществ, исключить этап очистки экстракта, что сокращает потери искомого вещества, ускоряет выпаривание экстрагента, повышает чувствительность и избирательность метода, сокращает продолжительность проведения анализа.
Использование 95% этанола для экстракции повышает чувствительность и избирательность способа. Экстракция 95% этанолом позволяет извлечь максимальное количество левомицетина, так как его растворимость в 95% этаноле выше, чем в этилацетате.
Фильтрация, обезвоживание экстракта сернокислым натрием сокращает продолжительность проведения анализа.
Использование для растворения сухого остатка этилацетата повышает избирательность метода. Кроме того, использование для растворения сухого остатка этилацетата менее опасно, чем использование метанола.
Использование жидкостного хроматографа с детектором спектрофотометрическим UVV 104М и колонкой Диасфер-110С-16 (150×4) мм, с размером частиц сорбента 5 мкм, а также элюента, состоящего из смеси ацетонитрил-вода-диэтиламин в соотношении 30:70:0,1 позволяет получить острый, симметричный пик левомицетина, коэффициент асимметрии которого близок к 1, а также сократить время удерживания в колонке, что повышает чувствительность, избирательность метода и сокращает время проведения анализа.
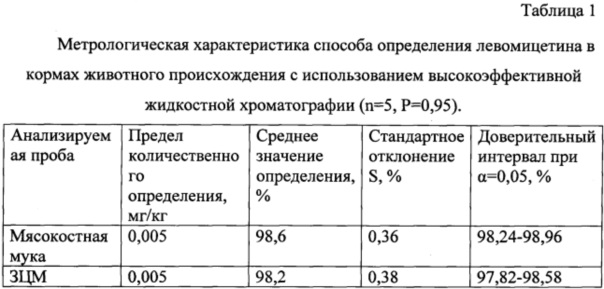
Способ определения левомицетина в кормах животного происхождения с использованием высокоэффективной жидкостной хроматографии осуществляют следующим образом. Берут, например, 250 мг мясокостной муки, гомогенизируют. Далее экстрагируют 95% этанолом по 5 мл трижды по 20 минут. Отдельные экстракты объединяют, фильтруют через воронку с бумажным фильтром, заполненным безводным сернокислым натрием, в грушевидную колбу для выпаривания, затем упаривают на ротационном испарителе при температуре водяной бани 40°С досуха. Полученный сухой остаток растворяют в 1 мл этилацетата и аликвоту вводят в жидкостный хроматограф «Хромос-ЖХ 301» с детектором спектрофотометрическим UVV 104М и колонкой Диасфер-110С-16 (150×4) мм, с размером частиц сорбента 5 мкм. Рабочая длина волны 278 нм. В качестве элюента используют подвижную фазу ацетонитрил-вода-диэтиламин в соотношении 30:70:01. Скорость потока 500 мкл/мин. Время удерживания левомицетина 5,305 мин. Нижний предел обнаружения 0,005 мг/кг. Проводят обработку результатов анализа. Количество левомицетина в пробе корма животного происхождения (X, мг/кг) рассчитывают по формуле
где X — содержание левомицетина в пробе, мг/кг;
А — количество левомицетина в стандартном растворе, введенном в хроматограф, нг;
S1 — площадь пика стандартного раствора левомицетина, введенного в хроматограф, мм 2 ;
S2 — площадь пика левомицетина в пробе, мм 2 ;
V1 — объем экстракта, введенный в хроматограф, мкл;
V2 — объем экстракта после растворения сухого оcтатка, мл;
Р — навеска анализируемого образца, г.
Метрологическая характеристика способа определения левомицетина в кормах животного происхождения с использованием высокоэффективной жидкостной хроматографии представлена в таблице 1.
Анализ метрологических характеристик свидетельствует о высокой чувствительности и избирательности предлагаемого способа.
Предлагаемый способ определения левомицетина в кормах животного происхождения с использованием высокоэффективной жидкостной хроматографии обеспечивает повышение чувствительности и избирательности за счет уменьшения навески пробы; использования в качестве экстрагента 95% этанола; использования жидкостного хроматографа с детектором спектрофотометрическим UVV 104М и колонкой Диасфер-110С-16 (150×4) мм, с размером частиц сорбента 5 мкм, а также элюента, состоящего из смеси ацетонитрил-вода-диэтиламин в соотношении 30:70:01. Чувствительность предлагаемого способа определения левомицетина повышается в 2 раза.
Предлагаемый способ определения левомицетина в кормах животного происхождения с использованием высокоэффективной жидкостной хроматографии обеспечивает сокращение длительности проведения анализа за счет уменьшения массы пробы корма, проведения фильтрации, обезвоживания экстракта сернокислым натрием, использования жидкостного хроматографа с детектором спектрофотометрическим UVV 104М и колонкой Диасфер-110С-16 (150×4) мм, с размером частиц сорбента 5 мкм, а также элюента, состоящего из смеси ацетонитрил-вода-диэтиламин в соотношении 30:70:01. Длительность проведения анализа предлагаемым способом сокращается в 1,8 раза.
Заявляемый способ определения левомицетина в кормах животного происхождения с использованием высокоэффективной жидкостной хроматографии может быть использован в лабораториях для контроля качества кормов, а также для оценки качества сырья животного происхождения, предназначенного для производства кормов, и определения содержания левомицетина в корме при добавлении в него антибиотика с целью групповой фармакотерапии.
Заявляемый способ определения левомицетина в кормах животного происхождения с использованием высокоэффективной жидкостной хроматографии апробирован в химико-токсикологическом отделе БУ ОО «Омская областная ветеринарная лаборатория».
2. Определение остаточных количеств левомицетина (Хлорамфеникола, Хлормицетина) в продуктах животного происхождения методом высокоэффективной жидкостной хроматографии и иммуноферментного анализа. Методические указания. МУК 4.1.1912 — 04 // Сборник методических документов, необходимых для обеспечения применения Федерального закона от 12 июня 2008 г. — №88 ФЗ «Технический регламент на молоко и молочную продукцию». — М.: Федеральный центр гигиены и эпидемиологии Роспотребнадзора, 2009. — С. 41-47 (прототип).
Способ определения левомицетина в кормах животного происхождения с использованием высокоэффективной жидкостной хроматографии, включающий отбор пробы, гомогенизацию, экстракцию, упаривание, растворение сухого остатка, введение растворенного сухого остатка в хроматограф с использованием элюента, обработку результатов анализа, отличающийся тем, что в качестве пробы берут навеску кормов животного происхождения массой от 200 до 500 мг, экстракцию проводят 95% этанолом, перед упариванием экстракт фильтруют, обезвоживают сернокислым натрием, растворение сухого остатка проводят в этилацетате, в качестве хроматографа используют жидкостной хроматограф с детектором спектрофотометрическим UVV 104М и колонкой Диасфер–110C-16 (150×4) мм, с размером частиц сорбента 5 мкм, в качестве элюента используют смесь ацетонитрил-вода-диэтиламин в соотношении 30:70:0,1.
источник
Владельцы патента RU 2431829:
Группа изобретений относится к концентрированию и определению антибиотика левомицетина в пищевых продуктах методом обращенно-фазовой высокоэффективной жидкостной хроматографии. Сущность изобретения: левомицетин экстрагируют из пробы ацетонитрилом, отделяют на центрифуге раствор, в котором находится антибиотик, к центрифугату добавляют сорбент — вермикулит при массовом соотношении вермикулита, равном 0,1 г сорбента на 1 г жира в пробе, и встряхивают в течение 30 минут, затем раствор отделяют и пропускают через смесь сорбентов — активированного угля и вермикулита, взятых в массовом соотношении, равном 1:1, и промывают дистиллированной водой, элюируют левомицетин этанолом, элюат упаривают досуха на водяной бане и охлаждают до комнатной температуры, затем остаток растворяют в подвижной фазе (ацетонитрил:вода, 70:30 об./об.), раствор левомицетина переносят дозатором в сухие виалы и количественно определяют левомицетин методом ОФ ВЭЖХ с УФ-детектированием при длине волны 278 нм; содержание антибиотика рассчитывают при помощи градуировочного графика. Достигается повышение точности и безопасности анализа. 2 н.п. ф-лы, 1 табл., 4 ил.
Изобретение относится к аналитической химии, а именно к способам определения антибиотика левомицетина в пищевых продуктах методом обращенно-фазовой высокоэффективной жидкостной хроматографии.
Антибиотики, в частности левомицетин (хлорамфеникол), используют в качестве эффективных противоинфекционных средств в ветеринарии и при профилактике заболеваний домашней птицы и скота. При этом антибиотик способен переходить в мясо, молоко животных, яйца птиц и другие пищевые продукты. Так как в последние годы в мире наблюдается устойчивая тенденция ужесточения требований к качеству пищевых продуктов, необходима разработка и введение в практику новых, более эффективных и чувствительных методов анализа.
Существуют различные методы определения левомицетина в пищевых продуктах (иммуноферментный анализ, химические методы (качественные реакции на данный антибиотик с различными специально подобранными реагентами), физико-химические методы (вольтамперометрия, полярография, спектрофотометрия), тонкослойная хроматография, газовая хроматография). При использовании данных методов анализа часто сталкиваются с определенными трудностями, например необходимостью использования специфических, редких и достаточно дорогостоящих реактивов; невозможностью определения сразу нескольких препаратов при совместном присутствии и неудовлетворительным нижним пределом обнаружения антибиотика; необходимостью использования предколонки (для газовой хроматографии).
Одним из наиболее перспективных методов определения антибиотика левомицетина является метод обращенно-фазовой высокоэффективной жидкостной хроматографии (ОФ ВЭЖХ), который дает возможность извлекать, разделять, идентифицировать и количественно определять лекарственные препараты в биологических жидкостях.
Известен способ количественного определения левомицетина в пищевых продуктах измерением массовой доли антибиотика методом ОФ ВЭЖХ с фотометрическим детектированием с использованием жидкостного хроматографа «Люмахром» [1]. Способ состоит в экстракции левомицетина из образца дистиллированной водой, дальнейшей очистке экстракта при помощи концентрирующего патрона ДИАПАК-C16 и в определении левомицетина методом ОФ ВЭЖХ с использованием фотометрического детектора (254 нм). Патроны ДИАПАК-С16 представляют собой корпусы из химически устойчивого полимера, заполненные высококачественными химически модифицированными сорбентами на основе силикагеля с привитыми гексадецильными группами. Способ имеет ряд недостатков: левомицетин из проб экстрагируется водой, что затрудняет дальнейший анализ, т.к. экстракты получаются достаточно загрязненными посторонними, мешающими определению антибиотика веществами; способ предполагает использование летучих и токсичных растворителей — хлороформа и гексана; определение левомицетина методом ОФ ВЭЖХ с ультрафиолетовым детектором проводят при длине волны 254 нм. Однако в этой области спектра интенсивное поглощение характерно для практически всех веществ, содержащих ароматический цикл; следовательно, известный метод определения левомицетина не гарантирует точных результатов.
Исследована возможность патронов ДИАПАК-C16 для концентрирования антибиотиков цефазолина и левомицетина из разбавленных растворов с последующим их определением методом ОФ-ВЭЖХ [2]. Сорбцию препаратов проводят в статическом и динамическом режимах, антибиотики десорбируют ацетонитрилом, полученные растворы анализируют методом ОФ ВЭЖХ с использованием подвижной фазы (ацетонитрил-вода, 50:50 об./об.); колонки Zorbax ODS C18 (15×0,46 см), рабочие длины волн — 210 и 254 нм. Однако данная методика позволяет осуществлять выделение и анализ антибиотиков из разбавленных водных растворов фармацевтических препаратов и не адаптирована для анализа пищевых продуктов с высоким содержанием жира.
Известен способ определения левомицетина в мясе краба и в креветках с помощью ВЭЖХ с масс-детектированием [3]. Антибиотик экстрагируют этилацетатом, в качестве подвижной фазы используют метанол. Недостатками данного способа являются: применение высокотоксичного метанола в качестве подвижной фазы; использование дорогостоящего и сложного в освоении жидкостного хроматографа с масс-детектором, что затрудняет широкое применение известного способа анализа.
Наиболее близким по технической сущности и достигаемому результату к способу определения левомицетина в пищевых продуктах является способ извлечения антибиотиков из пищевой продукции органическими растворителями — метанолом и ацетонитрилом с последующей очисткой экстракта растворами Карреза (водный раствор Zn(СН3СОО)2 и K4[Fe(CN)6]×3H2O), после чего экстракт обрабатывают диэтиловым эфиром для удаления липидов и анализируют методом ОФ ВЭЖХ с использованием подвижной фазы (ацетонитрил:вода, 70:30 об./об.) (колонка Zorbax ODS C18 (15×0,46 см), с УФ-детектированием при 220, 254 и 273 нм) [4]. Количество антибиотика в пробе определяют с помощью градуировочного графика. Предел обнаружения антибиотика левомицетина в способе-прототипе составляет 0,9±0,2 нг в одной порции раствора, вводимой в хроматограф для анализа. Недостатками данного способа являются необходимость применения высокотоксичного реагента — метанола, а также недостаточная точность определения левомицетина вследствие частичного осаждения антибиотика на хлопьевидном осадке гексацианоферрата цинка.
Задачей заявляемого изобретения является разработка воспроизводимого, прецизионного способа выделения и концентрирования левомицетина из пищевых продуктов, а также его количественного определения.
Поставленная задача достигается тем, что левомицетин экстрагируют из пробы ацетонитрилом, отделяют на центрифуге раствор, в котором находится антибиотик, к центрифугату добавляют сорбент — вермикулит (массовое соотношение которого 0,1 г сорбента на 1 г жира в пробе) и встряхивают в течение 30 минут, затем раствор отделяют и пропускают через смесь сорбентов — активированного угля и вермикулита, взятых в массовом соотношении, равном 1:1, и промывают дистиллированной водой, элюируют левомицетин этанолом, элюат упаривают досуха на водяной бане и охлаждают до комнатной температуры, затем остаток растворяют в подвижной фазе (ацетонитрил:вода, 70:30 об./об.), раствор левомицетина переносят дозатором в сухие виалы и количественно определяют левомицетин методом ОФ ВЭЖХ с УФ-детектированием при длине волны 278 нм; содержание антибиотика рассчитывают при помощи градуировочного графика.
Поставленная задача наилучшим образом решается новыми, ранее не известными из уровня техники условиями пробоподготовки образца, а именно:
— последовательным применением сорбентов:
— вначале — вермикулита, массовое соотношение которого 0,1 г сорбента на 1 г жира в пробе;
— затем — смеси активированного угля и вермикулита при их массовом соотношении, равном 1:1;
В заявляемом способе используют вермикулит (природный цеолит) Кокшаровского месторождения Приморского края.
Указанные существенные отличительные признаки заявляемого изобретения обеспечивают концентрирование антибиотика, очистку экстракта от мешающих анализу компонентов, что особенно актуально при анализе пищевых продуктов с высоким (>30%) содержанием жира, и позволяют определить левомицетин известным ОФ ВЭЖХ-методом с достаточной степенью точности.
Техническим результатом предлагаемого способа определения содержания левомицетина в пищевых продуктах является повышение точности количественного определения антибиотика в пищевых продуктах с различным содержанием жира, а также снижение токсичности проведения аналитических исследований.
Способ определения содержания левомицетина в пищевых продуктах осуществляют следующим образом. Левомицетин экстрагируют из пробы ацетонитрилом и отделяют раствор, в котором находится антибиотик с примесями, от осадка (свернувшийся белок). В полученный раствор антибиотика в ацетонитриле добавляют вермикулит, массовое соотношение которого 0,1 г сорбента на 1 г жира в пробе, встряхивают колбу со смесью в течение 30 минут на встряхивателе и отделяют раствор от сорбента. Отфильтрованный раствор пропускают через смесь сорбентов (активированный уголь и вермикулит, взятых при массовом соотношении, равном 1:1), промывают дистиллированной водой и элюируют антибиотик этанолом. Элюат в круглодонных колбах упаривают досуха на водяной бане, охлаждают до комнатной температуры и растворяют в подвижной фазе (ацетонитрил:вода, 70:30 об./об.). Раствор левомицетина переносят дозатором в сухие виалы и количественно определяют антибиотик методом ОФ ВЭЖХ с УФ-детектированием при длине волны 278 нм; содержание антибиотика рассчитывают при помощи градуировочного графика.
Экспериментально установлена необходимость последовательного применения сорбентов: вначале — вермикулита для дополнительной очистки экстракта от примесей. Необходимость использования вермикулита при его массовом соотношении, равном 0,1 г сорбента на 1 г жира в пробе, подтверждена экспериментальным путем и обусловлена тем, что при статической сорбции из раствора на вермикулите происходит сорбция примесей (липиды, белки и др.), которые присутствуют в экстракте. Таким образом, достигается предварительная очистка экстракта от балластных веществ.
Для сорбции левомицетина из раствора после отделения вермикулита используется бинарный сорбент (активированный уголь и вермикулит при их массовом соотношении, равном 1:1), применение которого позволяет не только проводить доочистку экстракта от соэкстрагированных примесей, но и концентрировать левомицетин в количествах, необходимых для хроматографического анализа. Активированный уголь сорбирует примеси из раствора левомицетина в этаноле, обеспечивает получение прозрачного и пригодного для ОФ ВЭЖХ-анализа раствора левомицетина. Для того чтобы минимизировать возможность сорбции антибиотика на угле, необходимое количество активированного угля предварительно рассчитывают по формуле (1), если содержание жира в продукте ≥3,9%; и по формуле (2), если содержание жира в продукте меньше чем 3,9%:
mR — масса остатка после экстракции ацетонитрилом;
f — содержание жира в исследуемом продукте.
где mS2 — масса сорбента, необходимая для анализа пищевого продукта, с содержанием жира менее 3,9%,
f — содержание жира в исследуемом продукте.
После расчета навески активированного угля, необходимой для анализа продукта на левомицетин, готовят смесь сорбентов при их массовом соотношении, равном 1:1; такое соотношение сорбентов является оптимальным и установлено экспериментально.
Изменение соотношения активированный уголь:вермикулит приводит либо к уменьшению степени извлечения антибиотика, либо к получению мутных растворов, непригодных для анализа методом ОФ ВЭЖХ.
Элюирование левомицетина осуществляется этанолом, поскольку этот растворитель обладает достаточной полярностью для наиболее полного элюирования левомицетина с сорбента. Менее полярные растворители извлекают с сорбента и неполярные примеси, что приводит к уменьшению точности анализа антибиотика заявляемым способом. Не маловажным при выборе растворителя на стадии элюирования левомицетина является меньшая токсичность этанола по сравнению с метанолом, используемым в способе-прототипе.
Заявляемый способ впервые позволяет анализировать пищевые продукты с различным содержанием жира — от 1,0% до 60% с достаточно высокой степенью извлечения антибиотика, а именно (98,5-90)%, что подтверждается данными таблицы.
Степень извлечения левомицетина из продуктов с различным содержанием жира
Исследуемый продукт
Жирность исследуемого продукта, %
Степень извлечения, %
Маргарин 1
60,0
88,9
89,3
Маргарин 2
40,0
91,2
90,9
Молоко
3,9
96,4
97,1
Яйца куриные
1,2
98,7
98,9
Правильность предлагаемого способа определения содержания левомицетина в пищевых продуктах подтверждена методом «введено-найдено» и экспериментальными данными (примеры 1-3); хроматограммы представлены на фигурах 1-4.
На фиг.1 представлена хроматограмма стандартного раствора левомицетина с концентрацией антибиотика 0,01 мг/мл, приготовленного из фармацевтического препарата «Хлорамфеникола натрия сукцинат», приобретенного в аптечной сети. Пик вещества со временем удерживания 2,492 мин принят за пик левомицетина.
На фиг.2 представлена хроматограмма раствора левомицетина с концентрацией 0,01 мг/мл, внесенного в пищевой продукт (растительный маргарин, жирностью 40%) и выделенного из него по заявляемому способу. Пик вещества со временем удерживания 2,429 мин принят за пик левомицетина.
На фиг.3 представлена хроматограмма раствора левомицетина, выделенного по заявляемому способу из пищевого продукта «яйцо куриное». Пик вещества со временем удерживания 2,418 мин принят за пик левомицетина.
На фиг.4 представлена хроматограмма раствора левомицетина с концентрацией 0,01 мг/мл, внесенного в пищевой продукт (растительный маргарин, жирностью 60%) и выделенного из него по заявляемому способу. Пик вещества со временем удерживания 2,437 мин принят за пик левомицетина.
Возможность осуществления заявляемого способа иллюстрируется примерами.
Пример 1. Определение левомицетина (хлорамфеникола) в яйцах куриных
В коническую колбу вместимостью 100,0 мл вносят 50 г куриных яиц, предварительно тщательно гомогенизированных в стеклянной посуде, приливают 100 мл ацетонитрила (хч) и ставят на встряхиватель на 2 часа. Полученный экстракт фильтруют через стекловату, помещенную в стеклянную воронку и промытую 5 мл ацетонитрила. В полученный раствор добавляют 1 г вермикулита. Колбу с экстрактом и сорбентом помещают на встряхиватель на 30 минут, фильтруют раствор и пропускают через бинарный сорбент — смесь 3,85 г активированного угля и 3,85 г вермикулита, после чего сорбенты промывают 10 мл дистиллированной воды. Затем левомицетин элюируют 10 мл этанола и упаривают раствор на водяной бане в круглодонной колбе досуха. Колбу охлаждают на воздухе до комнатной температуры и ополаскивают 1 мл подвижной фазы (ацетонитрил:вода, 70:30 об./об.). Раствор антибиотика в подвижной фазе собирают дозатором на 1000 мкл и переносят в сухую чистую виалу. Количество антибиотика находят методом ОФ ВЭЖХ (жидкостный хроматограф Shimadzu LC-6A с УФ-детектором, колонка Zorbax ODS C18 (15×0,46 см), подвижная фаза (ацетонитрил:вода, 70:30 об./об.) при рабочей длине волны 278 нм. Содержание левомицетина (мг/кг), рассчитанное по методу градуировочного графика составило: 0,0140±0,0011 мг/кг; степень извлечения — 98,9% (фиг.3).
Пример 2. Определение левомицетина в маргарине с жирностью 40,0%
Определение антибиотика проводили по примеру 1, но перед экстракцией 50 г маргарина растопили в колбе при температуре 55°С (достаточная температура, чтобы растопить жир, содержащийся в маргарине, и не разрушить антибиотик), добавили 1 мл раствора левомицетина с концентрацией 0,1 мг/мл. Затем, не охлаждая образец, прибавили 100 мл ацетонитрила и проводили экстракцию левомицетина на встряхивателе в течение 3 часов при температуре 50°С. В полученный раствор добавили 1 г вермикулита. Колбу с экстрактом и сорбентом помещали на встряхиватель на 30 минут, отфильтровывали раствор и пропускали через бинарный сорбент — смесь 27,0 г активированного угля и 27,0 г вермикулита. Далее — по примеру 1. Содержание левомицетина составило 0,09105 мг/кг; степень извлечения — 91% (фиг.2).
Пример 3. Определение содержания левомицетина в маргарине жирностью 60,0% Количество внесенного антибиотика такое же, как в примере 2. Время экстракции ацетонитрилом — 4 часа. Бинарный сорбент — 40,5 г активированного угля и 45,5 г вермикулита. Содержание левомицетина составило 0,0891 мг/кг; степень извлечения — 89% (фиг.4).
Экспериментально установлено, что в зависимости от содержания жира в продукте время экстракции необходимо изменять. Для продуктов с содержанием жира от 1 до 10% оно составляет 2 часа, для продуктов с содержанием жира от 10 до 40% — 3 часа, выше 40% — 4 часа. Увеличение времени экстракции более 4 часов не приводит к увеличению степени извлечения антибиотика; при времени экстракции менее 2 часов экстракция левомицетина не является полной.
Заявляемый способ выделения, концентрирования и идентификации антибиотика левомицетина (хлорамфеникола) прост, не трудоемок, не требует большого количества реактивов и может быть применен в любой химической лаборатории, где имеется жидкостный хроматограф с УФ-детектором. Погрешность измерения составляет 3-4% для концентрации до 0,01 мг/кг и 10% для больших концентраций.
1. № М 01-28-2002. «Методика выполнения измерений массовой доли левомицетина (хлорамфеникола) в продуктах животного происхождения методом ОФ ВЭЖХ с фотометрическим детектированием с использованием жидкостного хроматографа «Люмахром»». — г.Санкт-Петербург: ООО «Люмэкс», 2008.
2. Концентрирование антибиотиков цефазолина, цефотаксима и левомицетина на модифицированных кремнеземах. / Л.И.Соколова, И.В.Чучалина. // Журнал аналитической химии. — 2006. — Т.61, №12, с.1238-1242.
3. Rupp. H.S. Liquid chromatography/tandem mass spectrometry analysis of chloramphenicol in cooked crabmeat / H.S.Rupp, J.S.Stuart, J.A.Hurlbut // Journal of AOAC International. — 2005. — Vol.88, N 4. — PP.1155-1159.
4. Определение бензилпенициллина, левомицетина и тетрациклина в пищевых продуктах методом высокоэффективной жидкостной хроматографии. / Л.И.Соколова, А.П.Черняев. // Журнал аналитической химии. — 2001. — Т.56, №11, с.1177-1180.
1. Способ определения содержания левомицетина в пищевых продуктах, включающий экстракцию левомицетина из пробы ацетонитрилом, отделение от осадка раствора, содержащего левомицетин, очистку раствора сорбентом-вермикулитом при массовом соотношении 0,1 г сорбента на 1 г жира в пробе, доочистку раствора и концентрирование из него левомицетина при пропускании через сорбент в виде смеси активированного угля и вермикулита при их массовом соотношении 1:1, десорбцию левомицетина при элюировании этанолом, упаривание элюата с последующим растворением охлажденного остатка в подвижной фазе ацетонитрил-вода при их объемном соотношении 70:30, анализ полученного раствора методом обращенно-фазовой высокоэффективной жидкостной хроматографии и расчет содержания левомицетина при помощи градуировочного графика.
2. Сорбент для осуществления способа по п.1, содержащий активированный уголь и вермикулит при их массовом соотношении, равном 1:1.
источник
научная статья по теме ОПТИМИЗАЦИЯ УСЛОВИЙ ОПРЕДЕЛЕНИЯ ОСТАТОЧНЫХ КОЛИЧЕСТВ ЛЕВОМИЦЕТИНА (ХЛОРАМФЕНИКОЛА) В КОРОВЬЕМ МОЛОКЕ МЕТОДОМ ОБРАЩЕННО-ФАЗОВОЙ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ Химия
Авторы работы:
Научный журнал:
Текст научной статьи на тему «ОПТИМИЗАЦИЯ УСЛОВИЙ ОПРЕДЕЛЕНИЯ ОСТАТОЧНЫХ КОЛИЧЕСТВ ЛЕВОМИЦЕТИНА (ХЛОРАМФЕНИКОЛА) В КОРОВЬЕМ МОЛОКЕ МЕТОДОМ ОБРАЩЕННО-ФАЗОВОЙ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ»
ЖУРНАЛ АНАЛИТИЧЕСКОЙ ХИМИИ, 2013, том 68, № 2, с. 203-207
ОПТИМИЗАЦИЯ УСЛОВИЙ ОПРЕДЕЛЕНИЯ ОСТАТОЧНЫХ КОЛИЧЕСТВ ЛЕВОМИЦЕТИНА (ХЛОРАМФЕНИКОЛА) В КОРОВЬЕМ МОЛОКЕ МЕТОДОМ ОБРАЩЕННО-ФАЗОВОЙ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ
Ярославский государственный университет им. П.Г. Демидова, факультет биологии и экологии
150000Ярославль, пр-д Матросова, 9 Поступила в редакцию 27.02.2012 г., после доработки 05.07.2012 г.
Разработана чувствительная, селективная, точная и надежная методика определения остаточных количеств левомицетина (хлорамфеникола) в сыром и пастеризованном коровьем молоке, включающая фильтрование образца, очистку образца твердофазной экстракцией, очистку элюата жидкостной экстракцией, концентрирование упариванием под вакуумом и определение обращенно-фазовой ВЭЖХ с детектированием при 278 нм. Подвижная фаза — смесь (15 : 85, по объему) ацетонитрила и воды, содержащей 1 об. % пропанола-2. Предел обнаружения 0.45 мкг/кг, предел количественного определения 1.40 мкг/кг, предел повторяемости 6.93%.
Левомицетин — антибиотик, активный в отношении как грамположительных, так и грамотри-цательных бактерий и некоторых вирусов [1, 2]. Жесткий контроль содержания левомицетина в пищевых продуктах обусловлен широким спектром побочных воздействий на организм человека. Систематическое поступление этого антибиотика в организм может вызывать дисбактериозы, аллергические реакции, нарушать микрофлору кишечника, деятельность некоторых ферментов. Длительный прием способствует распространению устойчивых форм микроорганизмов. Следует также учитывать вероятность негативного влияния левомицетина на ряд технологических процессов при изготовлении кисломолочных продуктов [2]. Угнетение микроорганизмов закваски может изменить их органолептические и физико-химические свойства. Антимикробный эффект левомицетина вызван способностью подавлять синтез белка, присоединяясь к А-участку 508 субъединицы бактериальной рибосомы [3, 4].
В соответствии с Федеральным законом № 88 «Технический регламент на молоко и молочную продукцию» и СанПиН 2.3.2.1078-01 использование левомицетина в молочных хозяйствах запрещено, а его содержание в молоке и молочных продуктах допускается до 10 мкг/кг [5, 6]. Исходя из этого, методика идентификации и определе-
ния должна обладать достаточно высокой чувствительностью, чтобы обнаруживать концентрацию аналита по крайней мере на уровне 5 мкг/кг, что соответствует половине регламентируемой ПДК. Более того, необходимо чтобы методика позволяла отделять анализируемое соединение от мешающих компонентов матрицы, т.е. была селективна. Не менее важна точность методики, которая позволяет не только определять концентрации левомицетина в образцах, близкие к истинному значению, но и исключать возможность получения ложноположительных и ложноотри-цательных результатов. Методика должна быть надежной, т.е. есть способной в течение длительного времени сохранять определенные параметры в необходимых пределах. Существующая методика определения лемомицетина в пищевых продуктах ВЭЖХ [2] отвечает этим требованиям только частично.
Существует ряд методов, позволяющих определять левомицетин в матрицах различного состава [2, 7—16]. Преимущественно это методы газовой и жидкостной хроматографии. Также применяют тонкослойную хроматографию (ТСХ) и иммуноферментный анализ (ИФА). ИФА [2] позволяет обнаружить левомицетин на достаточно низком уровне 0.08 мкг/кг, однако требует дорогостоящих реактивов, градуировки перед каждой
сериеи анализов и проведения испытания в течение короткого промежутка времени. Метод ТСХ [14] характеризуется высоким пределом обнаружения по сравнению с хроматографическими методами — 1.00 мкг/кг. Он требует длительной и трудоемкой пробоподготовки, включающей многократную жидкость-жидкостную экстракцию, центрифугирование, концентрирование, получение производных и сопровождается значительными потерями аналита.
Наиболее точными и чувствительными являются методы газовой и высокоэффективной жидкостной хроматографии [2, 7—13, 15]. При реализации методов газовой хроматографии требуется предварительное получение летучих силилиро-ванных производных левомицетина [15]. Сигнал регистрируют либо детектором электронного захвата (ДЭЗ) благодаря наличию двух атомов хлора в молекуле аналита, либо масс-селективным детектором (МСД). Пределы обнаружения при применении ДЭЗ у разных авторов находились в диапазоне 0.05-1.00 мкг/кг, для МСД — 0.02-0.60 мкг/кг [15].
Описанные методики ВЭЖХ [2, 7-13, 15, 16] выполняли на обращенно-фазовом сорбенте с МСД и детектированием в УФ-области спектра. При определении нет необходимости в получении производных, однако использование спектро-фотометрического (СФД) или диодно-матричного (ДМД) детекторов требует многоступенчатой пробоподготовки из-за их низкой селективности. МСД решает проблему селективности, одновременно повышая чувствительность, однако его стоимость часто достигает уровня полноценной системы для жидкостной хроматографии. Пределы обнаружения для СФД и ДМД находились в диапазоне 0.65-50.00 мкг/кг, для МСД — 0.003-0.100 мкг/кг [2, 7-13, 15, 16].
Цель данной работы — разработка чувствительной, селективной, точной, надежной, соответствующей современным требованиям нормативных документов [5, 6], методики определения остаточных количеств левомицетина в сыром и пастеризованном коровьем молоке методом об-ращенно-фазовой ВЭЖХ.
возможностью смешивания до трех компонентов подвижной фазы, автоматическим пробоотборником с диапазоном ввода образца от 0.1 до 100 мкл, термостатом колонок, спектрофотометрическим детектором, позволяющим регистрировать аналитические сигналы на четырех длинах волн одновременно, и хроматографической рабочей станцией Chromeleon 6.80. УФ-спектр спиртового раствора левомицетина получали на спектрофотометре UNICO 2802PC.
Подготовка образцов. Молоко является сложной коллоидной полидисперсной системой, в состав которой входит до 200 различных химических соединений [17] как органической, так и неорганической природы, что требует тщательной очистки образца от мешающих компонентов и выбора условий разделения. Образцы молока отбирались согласно ГОСТ 26809-86 [18]. Образец пастеризованного молока или молока-сырья предварительно дважды пропускали через слой гигроскопической ваты. Для анализа отбирали 10 мл пробы и пропускали через предварительно подготовленный патрон Диапак С16. Белки и гидрофильные соединения элюировали 5 мл дистиллированной воды, жир и другие гидрофобные компоненты удаляли 5 мл гексана. Левоми-цетин с патрона элюировали 15 мл хлороформа, элюат упаривался в роторном испарителе досуха при температуре водяной бани не выше 45°C, концентрат растворяли в 1 мл смеси ацетонитри-ла, воды и изопропанола и очищали от остатков жира и гидрофобных соединений двукратной экстракцией по 1 мл гексана. Полученный раствор хроматографировали.
Для оценки степени извлечения левомицетина к 9 мл образца добавляли 1 мл водного раствора левомицетина с концентрацией 100 мкг/л (предельная допустимая концентрация левомицетина составляет 10 мкг/л). Образец с добавкой подвергали всем этапам пробоподготовки и хроматогра-фировали. Степень извлечения рассчитывали по формуле:
Реактивы. В качестве стандартного образца использовали левомицетин РСО 9348-152-00494189 (99.15% основного вещества). В работе применяли ацетонитрил ос.ч., 3 сорт (НПК «Криохром»), пропанол-2, н-гексан, хлороформ х.ч. (ЗАО «Экос-1»), спирт этиловый ректификованный из пищевого сырья «Экстра» по ГОСТ Р 51652.
Оборудование. Хроматографическое определение проводили на хроматографической системе иШМа1е-3000 фирмы Dionex, укомплектованной вакуумным дегазатором, градиентным насосом с
где сх — концентрация левомицетина в образце с добавкой, полученная по градуировочному графику (мкг/л); cst — концентрация раствора левомицетина, введенного в образец (100 мкг/л).
Условия хроматографического определения:
предколонка (10 х 2.1 мм) Acclaim 120 (С18-фаза, 5 мкм, 120 А); колонка (150 х 2.1 мм) Acclaim 120 (С18-фаза, 3 мкм, 120 А); подвижная фаза ацетонитрил — 1.0%-ный (по объему) водный раствор пропанола-2 в объемном соотношении (15 : 85), режим элюирования изократический; расход подвижной фазы 0.2 мл/мин; температура термостата
ОПТИМИЗАЦИЯ УСЛОВИЙ ОПРЕДЕЛЕНИЯ ОСТАТОЧНЫХ КОЛИЧЕСТВ
колонок 30.0°С; объем пробы 40 мкл; длина волны детектора 278 нм, контроль при 254 нм.
Время удерживания левомицетина 27.7—28.6 мин.
Определение проводили методом внешнего стандарта. Из спиртового раствора левомицетина с концентрацией 100 мг/л готовили рабочий стандартный водный раствор с концентрацией 1000 мкг/л. Растворы для построения градуиро-вочного графика с концентрациями 50, 100, 250, 500 и 750 мкг/л готовили разбавлением стандартного раствора в мерных колбах вместимостью 10 мл подвижной фазой. Градуировочные растворы и рабочий стандартный раствор хроматогра-фировали в тех же условиях, что и исследуемые образцы молока. Для построения градуировочно-го графика готовили три серии растворов (всего 18 точек). Градуировочный график описывается уравнением y = 0.0063 + 0.0062х, где y — площадь пика, х — концентрация, мкг/л, с коэффициентом корреляции r2 = 0.99991.
УФ-спектр спиртового раствора с концентрацией 10 мг/л снимали относительно этанола в диапазоне 380—190 нм с шагом сканирования 1 нм в кюветах с l = 10 мм.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЯ
Выбор условий подготовки образцов и хромато-графического анализа. Необходимый уровень чувствительности методики достигали путем концентрирования аналита на патроне для твердофазной экстракции из 10 мл образца, упариванием элюата, введением в хроматограф большого объема пробы (40 мкл) и регистрацией аналитического сигнала в максимуме поглощения левомицетина. Спектр поглощения аналита имеет два максимума поглощения — при 216 и 278 нм (рис. 1).
Для дальнейшего прочтения статьи необходимо приобрести полный текст. Статьи высылаются в формате PDF на указанную при оплате почту. Время доставки составляет менее 10 минут. Стоимость одной статьи — 150 рублей.
АЛТУХОВА А.А., ДУРИЦИН Е.П., КИМ А.В., КОРОЛЕВ В.А., ШОРМАНОВ В.К. — 2010 г.
источник
Загрязнение кормов для животных микотоксинами, активное использование антибактериальных средств в животноводстве, повышение требований к пищевой ценности и подлинности молока, определяет необходимость выбора оптимальных методов аналитического контроля качества и безопасности молока, учитывая при этом доступность и воспроизводимость методик, оперативность получения результатов, экономические и иные факторы.
Низкий уровень предельно-допустимой концентрации афлатоксина М1 (не более 0,0005 мг/л, в продуктах детского питания — не допускается), запрет присутствия антибиотиков в пищевых продуктах [1], необходимость маркировки количества витаминов в продуктах детского питания требует организации высоко-квалифицированных лабораторий на предприятиях молочной промышленности. Для проведения анализа физико-химическими методами, как правило нужна многостадийная очистка субстратов, которая занимает много времени, приводит к потерям анализируемого вещества и увеличению ошибки анализа. Наиболее распространенный метод контроля этих веществ — высокоэффективная жидкостная хроматография (далее ВЭЖХ) предполагает наличие дорогостоящего оборудования и использование реактивов высокой степени очистки, что существенно сказывается на конечной стоимости анализа.
В последнее десятилетие наблюдается быстрое внедрение иммунохимических методов анализа (преимущественно, иммуноферментного метода, далее — ИФА ) в лабораторную практику, связанное с усовершенствованием техники такого анализа и обусловленное потребностью в быстрых, чувствительных, специфичных, производительных и простых методах.
Метод ИФА (или ELISA) был предложен в начале 70-х годов тремя независимыми группами исследователей: Engvall и Perlmann в Швеции, van Weemen и Schuur в Нидерландах и Rubenstein с сотр. в США [2].
Отличительными особенностями метода является высокая чувствительность, специфичность, высокая производительность, экспрессность, возможность использования минимально очищенных экстрактов, сравнительно низкая стоимость оборудования.
Метод основан на специфическом воздействии антигена (загрязняющего вещества) и антитела (полученного против контролируемого вещества). Образующийся комплекс антиген-антитело после его мечения ферментом детектируется по цветной реакции с помощью простых измерительных приборов.
Для контроля молока, как в зарубежной практике, так и в России уже широко используется иммуноферментный анализ. Для подтверждения результатов анализа применяется метод иммуноаффинной очистки с последующей высокоэффективной жидкостной хроматографией.
Огромное значение для его дальнейшего распространения имеет серийное производство высококачественных компонентов для иммуноферментного анализа — антител и коньюгатов, а также комплектных тест-систем для ИФА, выпускающихся под контролем системы качества ISO9000
Большой спектр тест-систем представлен серией RIDASCREEN FAST, RIDASCREEN и RIDA (Таблица 1), с помощью которых исследование молока по показателям качества и безопасности может быть выполнено за короткое время [3], причем возможно как количественное определение, так и полуколичественный анализ при визуальном учете результатов.
Таблица 1. Некоторые серийно выпускающиеся тест-системы и иммуноаффинные колонки для анализа молока
Наименование тест-системы
Предел обнаружения
Время пробоподготовки /анализа
Иммуноаффинные колонки AFLAPREP M
Определяется ВЭЖХ
40 мин
Иммуноаффинные колонки RIDA Aflatoxin
Определяется ВЭЖХ
40 мин
RIDASCREEN® Афлатоксин М1, 96 опр.
Определение антибиотиков и других антибактериальных препаратов
RIDASCREEN® Левомицетин, 96 опр.
0,0125 мкг/кг
10 мин/3 часа
RIDASCREEN® Тетрациклин, 96 опр.
1,5 мкг/л
30 мин/1,5 часа
RIDASCREEN® Стрептомицин, 96 опр.
20 мкг/л
1 час / 3 часа
RIDASCREEN® Сульфамезин, 96 опр.
10 мкг/л
30 мин/3 часа
RIDASCREEN® FAST Сульфамезин, 48 опр.
10 мкг/л
20 мин/ 20 мин
RIDASCREEN® Нитрофураны, 96 опр.
0,1 мкг/л
2+16+ 2 ч/1.5 ч
RIDASCREEN® Фторхинолоны, 96 опр.
1 мкг/л
1 час 15 мин
RIDASCREEN® FAST Витамин В12, 48 опр.
0,5 мкг/л
30 мин/30 мин
Иммуноаффинная колонка EASI-EXTRACT Витамин В12
Пробоподготовка перед ВЭЖХ
20 мин
RIDASCREEN® FAST Фолиевая кислота, 48 опр.
10 мкг/л
30 мин/30 мин
RIDASCREEN® Биотин, 96 опр.
0,3 мкг/л
3 часа/ 1.5 часа
Тест-системы серии Vita® FAST для определения тиамина (В1), рибофлавина (В2), ниацина (В3), пантотеновой кислоты (В5), пиридоксина (В6), биотина (В7), фолиевой кислоты (В9), цианкобаламина (В12), инозитола, холина
Превышает требования СанПиН по пищевой ценности продуктов питания
48 часов
0,2 мкг/л
1 час/2.5 часа
RIDASCREEN® Казеин, 48 опр.
0,5% коровьего казеина
2 часа/2,5 часа
RIDASCREEN® CIS, 48 опр.
0,1% коровьего молока в козьем или овечьем молоке (сыре)
10 мин/1.5 часа
RIDA® Quick CIS, 25 опр.
0,5 % коровьего молока в козьем или овечьем молоке (сыре)
10 мин
RIDASCREEN® GIS, 48 опр.
0,1% козьего молока в овечьем молоке (сыре)
10 мин/1.5 часа
RIDASCREEN® Бета-лактоглобулин, 96 опр.
5 мг/л
30 мин/3 часа
Очень многие тест-системы RIDASCREEN FAST и RIDASCREEN уже включены в официальные методы Минздрава России, Управления ветеринарии Федерального агентства по сельскому хозяйству РФ и Российской академии сельскохозяйственных наук.
Так, например, разработаны «Методические рекомендации по экспресс-определению афлатоксина М1 в молоке, сухом молоке и сыре с помощью тест-системы «RIDASCREEN AFLATOXIN M1» МР №17/ФЦ3739, «Методические рекомендации по экспресс-определению афлатоксина М1 в молоке и сухом молоке с помощью тест-системы «RIDASCREEN FAST AFLATOXIN M1» 17ФЦ/3735, «Определение остаточных количеств левомицетина в продуктах животного происхождения методом высокоэффективной жидкостной хроматографии и иммуноферментного анализа» МУК 4.1.1912-04, «Методические указания по количественному определению антибактериальных препаратов в продовольственном сырье и продуктах питания животного происхождения методом конкурентного иммуноферментного анализа» МУК 5-1-14/1005
Для определения афлатоксина М1 в молоке и молочных продуктах международная организация по стандартизации и международная молочная федерация также рекомендуют метод конкурентного иммуноферментного анализа, соответственно, ISO 14675: 2003 и IDF 186:2003
Кроме того, подготовлен стандартный метод ISO 14501:1998, использующийся для исследования молока и сухого молока на афлатоксин М1 с помощью метода иммуноафинной очистки (колонки AFLAPREP M, RIDA Aflatoxin), с последующей высокоэффективной жидкостной хроматографией со спектрофлуориметрической детекцией
Известно много вариантов постановки ИФА. Наибольшее распространение получил гетерогенный вариант иммуноферментного анализа, при котором антиген (определяемое соединение) или антитела фиксируется на твёрдой фазе, в качестве которой могут выступать полистироловый планшет, полистироловые бусины, пористая подложка или магнитный носитель
Для наглядности рассмотрим вариант гетерогенного конкурентного ИФА, реализованный в тест-системе RIDASCREEN FAST Афлатоксин М1
На рисунке 1 схематично показана лунка полистиролового планшета, в которой последовательно выполняются все стадии иммуноферментного анализа
Поставляемый в комплекте набора планшет сенсибилизирован «антителами захвата», наработанными к антителам к афлатоксину М1 (антигену)
Анализ выполняется следующим образом. Исследуемые или стандартные
растворы, препарат, содержащий антитела к афлатоксину М1 и препарат, содержащий коньюгат афлатоксина М1 с ферментом, дозируются в лунки планшета (см. Рисунок 2). При инкубации планшета в течение определенного времени молекулы афлатоксина М1 и молекулы коньюгата, конкурируя между собой, связываются антителами в объеме раствора. В то же самое время, при инкубации происходит иммуносорбция антител к определяемому антигену «антителами захвата» на поверхности лунок планшета (см. Рисунок 3).
На последующей стадии промывки из лунок планшета удаляются свободные молекулы коньюгата (см. Рисунок 4).
После промывки планшета в его лунки дозируется раствор, содержащий субстрат и хромоген. В процессе инкубации, при химическом взаимодействии субстрата с хромогеном, в котором ферментный фрагмент молекулы коньюгата, связанной на поверхности лунки, выступает в
качестве катализатора, образуются окрашенные продукты реакции (см. Рисунок 5).
После определенного времени развития данной цветной реакции, в результате которой хромоген окрашивается в голубой цвет, в лунки добавляется стоп-реагент, при этом голубой цвет раствора меняется на
Интенсивность окраски в лунках ИФА-планшета обратно пропорциональна концентрации афлатоксина М1, другими словами — чем насыщенней цвет растворов, тем меньше концентрация афлатоксина М1 в молоке (см. Рисунок 7)
Обработка результатов измерений может выполняться вручную, либо с помощью специализированного программного обеспечения, например, RIDA Soft.
Для выполнения исследований молока с помощью большинства тест-систем необходимы следующие материалы и оборудование:
ИФА-фотометр, снабженный фильтром на 450 нм
Одноканальные автоматические пипетки на 50 мкл и на 200 — 1000 мкл
8-канальные автоматические пипетки на 50 мкл и 250 мкл (рекомендуется)
Наконечники для автоматических пипеток
Стеклянная посуда
Центрифуга
Пробирки с притертыми стеклянными или винтовыми пробками
Рисунок 7. Калибровочная кривая, полученная для тест-системы RIDASCREEN FAST Афлатоксин М1
В заключение подчеркнем основные преимущества иммуноферментного метода анализа
сравнительно с другими методами:
Экспрессность и высокая производительность;
Простота пробоподготовки и анализа, удобство работы;
Высокая чувствительность и специфичность метода;
Гибкость метода — анализ может быть проведен по выбору вручную или автоматически;
Низкая стоимость требующегося оборудования и расходных материалов, небольшие затраты на постановку и поддержку метода;
Освоение метода не требует специальных знаний.
Литература
СанПиН 2.3.2.1078-01 Гигиенические требования безопасности и пищевой ценности пищевых продуктов. — М.: Минздрав России, 2002.
Иммунологические методы (под редакцией Г.Фримеля).1987г.; Стр.162, 163-170.
Галкин А. Теория и практика иммуноферментного анализа. — Био. — Июнь — 2003, с. 33.